<Back to Index>
- Element 95 Americium Am, 1944
- Chemist Glenn Theodore Seaborg, 1912
- Chemist Leon O. Morgan, 1900+
- Chemist Ralph A. James, 1900+
- Chemist Albert Ghiorso, 1915
PAGE SPONSOR

Americium is a transuranic radioactive chemical element that has the symbol Am and atomic number 95. This transuranic element of the actinide series is located in the periodic table below the lanthanide element europium, and thus by analogy was named after another continent, America.
Americium was first produced in 1944 by the group of Glenn T. Seaborg at the University of California, Berkeley. Although it is the third element in the transuranic series, it was discovered fourth, after the heavier curium. The discovery was kept secret and only released to the public in November 1945. Most americium is produced by bombarding uranium or plutonium with alpha particles in nuclear reactors – one tonne of spent nuclear fuel contains about 100 grams of americium. It is widely used in commercial ionization chamber smoke detectors, as well as in neutron sources and industrial gauges. Several unusual applications, such as a nuclear battery or fuel for space ships with nuclear propulsion, have been proposed for the isotope 242mAm, but they are as yet hindered by the scarcity and high price of this nuclear isomer.
Americium is a relatively soft radioactive metal with silvery white appearance. Its most common isotopes are 241Am and 243Am. In chemical compounds, they usually assume the oxidation state +3,
especially in solutions. Several other oxidation states are known,
which range from +2 to +7 and can be identified by their characteristic optical absorption spectra.
The crystal lattice of solid americium and its compounds contains
intrinsic defects, which are induced by self - irradiation with alpha
particles and accumulate with time; this results in a drift of some
material properties.
Although americium was likely produced in previous nuclear experiments, it was first intentionally synthesized, isolated and identified in late autumn 1944, at the University of California, Berkeley by Glenn T. Seaborg, Leon O. Morgan, Ralph A. James, and Albert Ghiorso. They used a 60 inch cyclotron at the University of California, Berkeley. The element was chemically identified at the Metallurgical Laboratory (now Argonne National Laboratory) of the University of Chicago. Following the lighter neptunium, plutonium, and heavier curium, americium was the fourth transuranium element to be discovered. At the time, the periodic table had been restructured by Seaborg to its present layout, containing the actinide row below the lanthanide one. This led to americium being located right below its twin lanthanide element europium; it was thus by analogy named after another continent, America: "The name americium (after the Americas) and the symbol Am are suggested for the element on the basis of its position as the sixth member of the actinide rare - earth series, analogous to europium, Eu, of the lanthanide series."
The new element was isolated from its oxides in a complex, multi - step process. First plutonium - 239 nitrate (239PuNO3) solution was coated on a platinum foil of about 0.5 cm2 area, the solution was evaporated and the residue was converted into plutonium dioxide (PuO2) by annealing. After cyclotron irradiation, the coating was dissolved with nitric acid, and then precipitated as the hydroxide using concentrated aqueous ammonia solution. The residue was dissolved in perchloric acid. Further separation was carried out by ion exchange, yielding a certain isotope of curium. The separation of curium and americium was so painstaking that those elements were initially called by the Berkeley group as pandemonium (from Greek for all demons or hell) and delirium (from Latin for madness).
Initial experiments yielded four americium isotopes: 241Am, 242Am, 239Am and 238Am. Americium - 241 was directly obtained from plutonium upon absorption of one neutron. It decays by emission of an α-particle to 237Np; the half-life of this decay was first determined as 510 ± 20 years but then corrected to 432.2 years.
![\mathrm{^{239}_{\ 94}Pu\ \xrightarrow {(n,\gamma)} \ ^{240}_{\ 94}Pu\ \xrightarrow {(n,\gamma)} \ ^{241}_{\ 94}Pu\ \xrightarrow [14.35 \ yr]{\beta^-} \ ^{241}_{\ 95}Am\ \left(\ \xrightarrow [432.2 \ yr]{\alpha} \ ^{237}_{\ 93}Np \right)}](aux1.png)
- The times are half-lives
The second isotope 242Am was produced upon neutron bombardment of the already created 241Am. Upon rapid β-decay, 242Am converts into the isotope of curium 242Cm (which had been discovered previously). The half-life of this decay was initially determined at 17 hours, which was close to the presently accepted value of 16.02 h.
![\mathrm{^{241}_{\ 95}Am\ \xrightarrow {(n,\gamma)} \ ^{242}_{\ 95}Am\ \left(\ \xrightarrow [16.02 \ h]{\beta^-} \ ^{242}_{\ 96}Cm \right)}](aux2.png)
The discovery of americium and curium in 1944 was closely related to the Manhattan Project;
the results were confidential and declassified only in 1945. Seaborg
leaked the synthesis of the elements 95 and 96 on the U.S. radio show
for children, the Quiz Kids, five days before the official presentation at an American Chemical Society meeting
on November 11, 1945, when one of the listeners asked whether any new
transuranium element beside plutonium and neptunium had been discovered
during the war. After the discovery of americium isotopes 241Am and 242Am, their production and compounds were patented listing only Seaborg as the inventor. The
initial americium samples weighed a few micrograms; they were barely
visible and were identified by their radioactivity. The first
substantial amounts of metallic americium weighing 40 – 200 micrograms
were not prepared until 1951 by reduction of americium (III) fluoride with barium metal in high vacuum at 1100 °C.
The longest lived and most common isotopes of americium, 241Am and 243Am, have half-lives of 432.2 and 7,370 years, respectively. Therefore, all primordial americium (americium that was present on Earth during its formation) should have decayed by now.
Existing americium is concentrated in the areas used for the atmospheric nuclear weapons tests conducted between 1945 and 1980, as well as at the sites of nuclear incidents, such as the Chernobyl disaster. For example, the analysis of the debris at the testing site of the first U.S. hydrogen bomb, Ivy Mike, (November 1, 1952, Enewetak Atoll), revealed high concentrations of various actinides including americium; due to military secrecy, this result was published only in 1956. Trinitite, the glassy residue left on the desert floor near Alamogordo, New Mexico, after the plutonium based Trinity nuclear bomb test on July 16, 1945, contains traces of americium - 241. Elevated levels of americium were also detected at the crash site of a US B-52 bomber, which carried four hydrogen bombs, in 1968 in Greenland.
In other regions, the average radioactivity due to residual americium is only about 0.01 picocuries (0.37 mBq). Atmospheric americium compounds are poorly soluble in common solvents and mostly adhere to soil particles. Soil analysis revealed about 1,900 higher concentration of americium inside sandy soil particles than in the water present in the soil pores; an even higher ratio was measured in loam soils.
Americium is produced mostly artificially in small quantities, for research purposes. A tonne of spent nuclear fuel contains about 100 grams of various americium isotopes, mostly 241Am and 243Am. Their prolonged radioactivity is undesirable for the disposal, and therefore americium, together with other long lived actinides, have to be neutralized. The associated procedure may involve several steps, where americium is first separated and then converted by neutron bombardment in special reactors to short lived nuclides. This procedure is well known as nuclear transmutation, but it is still being developed for americium.
A few atoms of americium can be produced by neutron capture reactions and beta decay in very highly concentrated uranium bearing deposits.
Americium has been produced in small quantities in nuclear reactors for decades, and kilograms of its 241Am and 243Am isotopes have been accumulated by now. Nevertheless, since it was first offered for sale in 1962, its price, about 1,500 USD per gram of 241Am, remains almost unchanged owing to the very complex separation procedure. The heavier isotope 243Am is produced in much smaller amounts; it is thus more difficult to separate, resulting in a higher cost of the order 100 – 160 USD/mg.
Americium is not synthesized directly from uranium – the most common reactor material – but from the plutonium isotope 239Pu. The latter needs to be produced first, according to the following nuclear process:
![\mathrm{^{238}_{\ 92}U\ \xrightarrow {(n,\gamma)} \ ^{239}_{\ 92}U\ \xrightarrow [23.5 \ min]{\beta^-} \ ^{239}_{\ 93}Np\ \xrightarrow [2.3565 \ d]{\beta^-} \ ^{239}_{\ 94}Pu}](aux3.png)
The capture of two neutrons by 239Pu (a so-called (n,γ) reaction), followed by a β-decay, results in 241Am:
![\mathrm{^{239}_{\ 94}Pu\ \xrightarrow {2(n,\gamma)} \ ^{241}_{\ 94}Pu\ \xrightarrow [14.35 \ yr]{\beta^-} \ ^{241}_{\ 95}Am}](aux4.png)
The plutonium present in spent nuclear fuel contains about 12% of 241Pu. Because it spontaneously converts to 241Am, 241Pu can be extracted and may be used to generate further 241Am. However, this process is rather slow: half of the original amount of 241Pu decays to 241Am after about 15 years, and the 241Am amount reaches a maximum after 70 years.
The obtained 241Am can be used for generating heavier americium isotopes by further neutron capture inside a nuclear reactor. In a light water reactor (LWR), 79% of 241Am converts to 242Am and 10% to its nuclear isomer 242mAm:
- 79%:
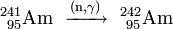
- 10%:
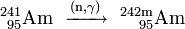
Americium - 242 has a half-life of only 16 hours, which makes its further up - conversion to 243Am, extremely inefficient. The latter isotope is produced instead in a process where 239Pu captures four neutrons under high neutron flux:
![\mathrm{^{239}_{\ 94}Pu\ \xrightarrow {4(n,\gamma)} \ ^{243}_{\ 94}Pu\ \xrightarrow [4.956 \ h]{\beta^-} \ ^{243}_{\ 95}Am}](aux7.png)
Most synthesis routines yield a mixture of different actinide isotopes in oxide forms, from which isotopes of americium need to be separated. In a typical procedure, the spent reactor fuel (e.g., MOX fuel) is dissolved in nitric acid, and the bulk of uranium and plutonium is removed using a PUREX type extraction (Plutonium –URanium EXtraction) with tributyl phosphate in a hydrocarbon. The lanthanides and remaining actinides are then separated from the aqueous residue (raffinate) by a diamide based extraction, to give, after stripping, a mixture of trivalent actinides and lanthanides. Americium compounds are then selectively extracted using multi - step chromatographicand centrifugation techniques with an appropriate reagent. A large amount of work has been done on the solvent extraction of americium. For example, a recent EU funded project codenamed "EUROPART" studied triazines and other compounds as potential extraction agents. Bis-triazinyl bipyridine complex has been recently proposed as such reagent as highly selective to americium (and curium). Separation of americium from the highly similar curium can be achieved by treating a slurry of their hydroxides in aqueous sodium bicarbonate with ozone, at elevated temperatures. Both Am and Cm are mostly present in solutions in the +3 valence state; whereas curium remains unchanged, americium oxidizes to soluble Am (IV) complexes which can be washed away.
Metallic americium is obtained by reduction from its compounds. Americium (III) fluoride was first used for this purpose. The reaction was conducted using elemental barium as reducing agent in a water- and oxygen - free environment inside an apparatus made of tantalum and tungsten.
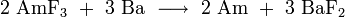
An alternative is the reduction of americium dioxide by metallic lanthanum or thorium:
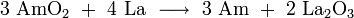
In the periodic table, americium is located right to plutonium, left to curium, and below the lanthanide europium, with which it shares many similarities in physical and chemical properties. Americium is a highly radioactive element. When freshly prepared, it has a silvery white metallic lustre, but then slowly tarnishes in air. With a density of 12 g/cm3, americium is lighter than both curium (13.52 g/cm3) and plutonium (19.8 g/cm3); but is heavier than europium (5.264 g/cm3) — mostly because of its higher atomic mass. Americium is relatively soft and easily deformable and has a significantly lower bulk modulus than the actinides before it: Th, Pa, U, Np and Pu. Its melting point of 1173 °C is significantly higher than that of plutonium (639 °C) and europium (826 °C), but lower than for curium (1340 °C).
At ambient conditions, americium is present in its most stable α form which has a hexagonal crystal symmetry, and a space group P63/mmc with lattice parameters a = 346.8 pm and c = 1124 pm, and four atoms per unit cell. The crystal consists of a double - hexagonal close packing with the layer sequence ABAC and so is isotypic with α-lanthanum and several actinides such as α-curium. The crystal structure of americium changes with pressure and temperature. When compressed at room temperature to 5 GPa, α-Am transforms to the β modification, which has a face - centered cubic (fcc) symmetry, space group Fm3m and lattice constant a = 489 pm. This fcc structure is equivalent to the closest packing with the sequence ABC. Upon further compression to 23 GPa, americium transforms to an orthorhombic γ-Am structure similar to that of α-uranium. There are no further transitions observed up to 52 GPa, except for an appearance of a monoclinic phase at pressures between 10 and 15 GPa. There is no consistency on the status of this phase in the literature, which also sometimes lists the α, β and γ phases as I, II and III. The β-γ transition is accompanied by a 6% decrease in the crystal volume; although theory also predicts a significant volume change for the α-β transition, it is not observed experimentally. The pressure of the α-β transition decreases with increasing temperature, and when α-americium is heated at ambient pressure, at 770 °C it changes into an fcc phase which is different from β-Am, and at 1075 °C it converts to a body - centered cubic structure. The pressure - temperature phase diagram of americium is thus rather similar to those of lanthanum, praseodymium and neodymium.
As with many other actinides, self damage of the crystal lattice due to alpha particle irradiation is intrinsic to americium. It is especially noticeable at low temperatures, where the mobility of the produced lattice defects is relatively low, by broadening of X-ray diffraction peaks. This effect makes somewhat uncertain the temperature of americium and some of its properties, such as electrical resistivity. So for americium - 241, the resistivity at 4.2 K increases with time from about 2 µOhm·cm to 10 µOhm·cm after 40 hours, and saturates at about 16 µOhm·cm after 140 hours. This effect is less pronounced at room temperature, due to annihilation of radiation defects; also heating to room temperature the sample which was kept for hours at low temperatures restores its resistivity. In fresh samples, the resistivity gradually increases with temperature from about 2 µOhm·cm at liquid helium to 69 µOhm·cm at room temperature; this behavior is similar to that of neptunium, uranium, thorium and protactinium, but is different from plutonium and curium which show a rapid rise up to 60 K followed by saturation. The room temperature value for americium is lower than that of neptunium, plutonium and curium, but higher than for uranium, thorium and protactinium.
Americium is paramagnetic in a wide temperature range, from that of liquid helium,
to room temperature, and above. This behavior is markedly different
from that of its neighbor curium which exhibit antiferromagnetic
transition at 52 K. The thermal expansion coefficient of americium is slightly anisotropic and amounts to (7.5 ± 0.2)×10−6/°C along the shorter a axis and (6.2 ± 0.4)×10−6/°C for the longer c hexagonal axis. The enthalpy of dissolution of americium metal in hydrochloric acid at standard conditions is −620.6 ± 1.3 kJ/mol, from which the standard enthalpy change of formation (ΔfH°) of aqueous Am3+ ion is −621.2 ± 2.0 kJ/mol−1. The standard potential Am3+/Am0 is 2.08 ± 0.01 V.
Americium readily reacts with oxygen and dissolves well in acids. The most common oxidation state for americium is +3, in which americium compounds are rather stable against oxidation and reduction. In this sense, americium is chemically similar to most lanthanides. The trivalent americium forms insoluble fluoride, oxalate, iodate, hydroxide, phosphate and other salts. Other oxidation states have been observed between +2 and +7, which is the widest range among the actinide elements. Their color in aqueous solutions varies as follows: Am3+ (colorless to yellow - reddish), Am4+ (yellow - reddish), AmVO2+; (yellow), AmVIO22+ (brown) and AmVIIO65- (dark green). All oxidation states have their characteristic optical absorption spectra, with a few sharp peaks in the visible and mid - infrared regions, and the position and intensity of these peaks can be converted into the concentrations of the corresponding oxidation states. For example, Am (III) has two sharp peaks at 504 and 811 nm, Am (V) at 514 and 715 nm, and Am (VI) at 666 and 992 nm.
Americium compounds with oxidation state +4 and higher are strong oxidizing agents, comparable in strength to the permanganate ion (MnO4-) in acidic solutions. Whereas the Am4+ ions are unstable in solutions and readily convert to Am3+, the +4 oxidation state occurs well in solids, such as americium dioxide (AmO2) and americium (IV) fluoride (AmF4).
All pentavalent and hexavalent americium compounds are complex salts such as KAmO2F2, Li3AmO4 and Li6AmO6, Ba3AmO6, AmO2F2. These high oxidation states Am (IV), Am (V) and Am (VI) can be prepared from Am (III) by oxidation with ammonium persulfate in dilute nitric acid, with silver (I) oxide in perchloric acid, or with ozone or sodium persulfate in sodium carbonate solutions. The pentavalent oxidation state of americium was first observed in 1951. It is present in aqueous solution in the form of AmO2+ ions (acidic) or AmO3- ions (alkaline) which are however unstable and subject to several rapid disproportionation reactions:
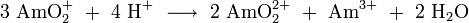
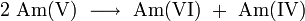
Two americium oxides are known, with the oxidation states +3 (Am2O3) and +4 (AmO2). Americium (III) oxide is a red brown solid with a melting point of 2205 °C. Americium (IV) oxide is the main form of solid americium which is used in nearly all its applications. As most other actinide dioxides, it is a black solid with a cubic (fluorite) crystal structure.
The oxalate of americium (III), vacuum dried at room temperature, has the chemical formula Am2(C2O4)3·7H2O. Upon heating in vacuum, it loses water at 240 °C and starts decomposing into AmO2 at 300 °C, the decomposition completes at about 470 °C. The initial oxalate dissolves in nitric acid with the maximum solubility of 0.25 g/L.
Halides of americium are known for the oxidation states +2, +3 and +4, where the +3 is most stable, especially in solutions.
Reduction of Am (III) compounds with sodium amalgam yields Am (II) salts – the black halides AmCl2, AmBr2 and AmI2. They are very sensitive to oxygen and oxidize in water, releasing hydrogen and converting back to the Am (III) state. Specific lattice constants are:
- Orthorhombic AmCl2: a = 896.3 ± 0.8 pm, b = 757.3 ± 0.8 pm and c = 453.2 ± 0.6 pm
- Tetragonal AmBr2: a = 1159.2 ± 0.4 and c = 712.1 ± 0.3 pm.
They can also be prepared by reacting metallic americium with an appropriate mercury halide HgX2, where X = Cl, Br or I:
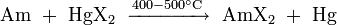
Americium (III) fluoride (AmF3) is poorly soluble and precipitates upon reaction of Am3+ and fluoride ions in weak acidic solutions:
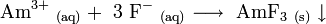
The tetravalent americium (IV) fluoride (AmF4) is obtained by reacting solid americium (III) fluoride with molecular fluorine:
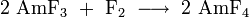
Another known form of solid tetravalent americium chloride is KAmF5. Tetravalent americium has also been observed in the aqueous phase. For this purpose, black Am(OH)4 was dissolved in 15-M NH3F with the americium concentration of 0.01 M. The resulting reddish solution had a characteristic optical absorption spectrum which is similar to that of AmF4 but differed from other oxidation states of americium. Heating the Am (IV) solution to 90 °C did not result in its disproportionation or reduction, however a slow reduction was observed to Am (III) and assigned to self irradiation of americium by alpha particles.
Most americium (III) halides form hexagonal crystals with slight variation of the color and exact structure between the halogens. So, chloride (AmCl3) is reddish and has a structure isotypic to uranium (III) chloride (space group P63/m) and the melting point of 715 °C. The fluoride is isotypic to LaF3 (space group P63/mmc) and the iodide to BiI3 (space group R3). The bromide is an exception with the orthorhombic PuBr3-type structure and space group Cmcm. Crystals of americium hexahydrate (AmCl3·6H2O) can be prepared by dissolving americium dioxide in hydrochloric acid and evaporating the liquid. Those crystals are hygroscopic and have yellow reddish color and a monoclinic crystal structure.
Oxyhalides of americium in the form AmVIO2X2, AmVO2X, AmIVOX2 and AmIIIOX can be obtained by reacting the corresponding americium halide with oxygen or Sb2O3, and AmOCl can also be produced by vapor phase hydrolysis:
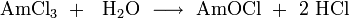
- The known chalcogenides of americium include the sulfide AmS2, selenides AmSe2 and Am3Se4, and tellurides Am2Te3 and AmTe2. The pnictides of americium (243Am) of the AmX type are known for the elements phosphorus, arsenic, antimony and bismuth. They crystallize in the rock - salt lattice.
Analogous to uranocene, americium forms an organometallic compound with two cyclooctatetraene ligands, that is (η8-C8H8)2Am. It also makes trigonal (η5-C5H5)3Am complexes with three cyclopentadienyl rings.
Formation of the complexes of the type Am(n-C3H7-BTP)3, where BTP stands for 2,6-di(1,2,4-triazin-3-yl)pyridine, in solutions containing n-C3H7-BTP and Am3+ ions has been confirmed by EXAFS. Some of these BTP type complexes selectively interact with americium and therefore are useful in its selective separation from lanthanides and another actinides.
Americium is an artificial element, and thus a biological function involving the element, like all elements heavier than tungsten, and thus all artificial elements, would be impossible. It has been proposed to use bacteria for removal of americium and other heavy metals from rivers and streams. Thus, Enterobacteriaceae of the genus Citrobacter precipitate americium ions from aqueous solutions, binding them into a metal - phosphate complex at their cell walls. Several studies have been reported on the biosorption and bioaccumulation of americium by bacteria and fungi.
The isotope 242m1Am (half-life 141 years) has the largest cross sections for absorption of thermal neutrons (5,700 barns), that results in a small critical mass for a sustained nuclear chain reaction. The critical mass for a bare 242m1Am sphere is about 9–14 kg (the uncertainty results from insufficient knowledge of its material properties). It can be lowered to 3–5 kg with a metal reflector and should become even smaller with a water reflector. Such small critical mass is favorable for portable nuclear weapons, but those based on 242m1Am are not known yet, probably because of its scarcity and high price. The critical masses of two other readily available isotopes,241Am and 243Am, are relatively high – 57.6 to 75.6 kg for 241Am and 209 kg for 243Am. Scarcity and high price yet hinder application of americium as a nuclear fuel in nuclear reactors.
There are proposals of very compact 10-kW high flux reactors using as little as 20 grams of 242m1Am. Such low power reactors would be relatively safe to use as neutron sources for radiation therapy in hospitals.
About 19 isotopes and 8 nuclear isomers are known for americium. There are two long lived alpha emitters, 241Am and 243Am with half-lives of 432.2 and 7,370 years, respectively, and the nuclear isomer 242m1Am has a long half-life of 141 years. The half-lives of other isotopes and isomers range from 0.64 microseconds for 245m1Am to 50.8 hours for 240Am. As with most other actinides, the isotopes of americium with odd number of neutrons have relatively high rate of nuclear fission and low critical mass.
Americium - 241 decays to 237Np emitting alpha particles of 5 different energies, mostly at 5.486 MeV (85.2%) and 5.443 MeV (12.8%). Because many of the resulting states are metastable, they also emit gamma rays with the discrete energies between 26.3 and 158.5 keV.
Americium - 242 is a short lived isotope with a half-life of 16.02 h. It mostly (82.7%) converts by β-decay to 242Cm, but also by electron capture to 242Pu (17.3%). Both 242Cm and 242Pu transform via nearly the same decay chain through 238Pu down to 234U.
Nearly all (99.541%) of 242m1Am decays by internal conversion to 242Am and the remaining 0.459% by α-decay to 238Np. The latter breaks down to 238Pu and then to 234U.
Americium - 243 transforms by α-emission into 239Np, which converts by β-decay to 239Pu, and the 239Pu changes into 235U by emitting an α-particle.
Americium is the only synthetic element to have found its way into the household, where one common type of smoke detector uses 241Am in the form of americium dioxide as its source of ionizing radiation. This isotope is preferred against 226Ra because
it emits 5 times more alpha particles and relatively little of harmful
γ-radiation. The amount of americium in a typical new smoke detector is
1 microcurie (37 kBq) or 0.28 microgram. This amount declines slowly as the americium decays into neptunium - 237, a different transuranic element with
a much longer half-life (about 2.14 million years). With its half-life
of 432.2 years, the americium in a smoke detector includes about 3% neptunium after 19 years, and about 5% after 32 years. The radiation passes through an ionization chamber, an air filled space between two electrodes, and permits a small, constant current between
the electrodes. Any smoke that enters the chamber absorbs the alpha
particles, which reduces the ionization and affects this current,
triggering the alarm. Compared to the alternative optical smoke
detector, the ionization smoke detector is cheaper and can detect
particles which are too small to produce significant light scattering;
however, it is more prone to false alarms.
As 241Am has a significantly longer half-life than 238Pu (432.2 years vs. 87 years), it has been proposed as an active element of radioisotope thermoelectric generators, for example in spacecraft. Although americium produces less heat and electricity – the power yield is 114.7 mW/g for 241Am and 6.31 mW/g for 243Am (cf. 390 mW/g for 238Pu) – and its radiation poses more threat to humans owing to neutron emission, the European Space Agency is planning to use americium for its space probes.
Another proposed space related application of americium is a fuel for space ships with nuclear propulsion. It relies on the very high rate of nuclear fission of 242mAm, which can be maintained even in a micrometer thick foil. Small thickness avoids the problem of self absorption of emitted radiation. This problem is pertinent to uranium or plutonium rods, in which only surface layers provide alpha particles. The fission products of 242mAm can either directly propel the spaceship or they can heat up a thrusting gas; they can also transfer their energy to a fluid and generate electricity through a magnetohydrodynamic generator.
One more proposal which utilizes the high nuclear fission rate of 242mAm
is a nuclear battery. Its design relies not on the energy of the
emitted by americium alpha particles, but on their charge, that is the
americium acts as the self sustaining "cathode". A single 3.2 kg 242mAm charge of such battery could provide about 140 kW of power over a period of 80 days. With all the potential benefits, the current applications of 242mAm are as yet hindered by the scarcity and high price of this nuclear isomer.
The oxide of 241Am pressed with beryllium is an efficient neutron source. Here americium acts as the alpha source, and beryllium produces neutrons owing to its large cross-section for the (α,n) nuclear reaction:
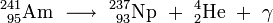
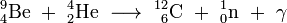
The most widespread use of 241AmBe neutron sources a neutron probe –
a device used to measure the quantity of water present in soil, as well
as moisture / density for quality control in highway construction. 241Am neutron sources are also used in well logging applications, as well as in neutron radiography, tomography and other radiochemical investigations.
Americium is a starting material for the production of other transuranic elements and transactinides – for example, 82.7% of 242Am decays to 242Cm and 17.3% to 242Pu. In the nuclear reactor, 242Am is also up - converted by neutron capture to 243Am and 244Am, which transforms by β-decay to 244Cm:
![\mathrm{^{243}_{\ 95}Am\ \xrightarrow {(n,\gamma)} \ ^{244}_{\ 95}Am\ \xrightarrow [10.1 \ h]{\beta^-} \ ^{244}_{\ 96}Cm}](eaux18.png)
Irradiation of 241Am by 12C or 22Ne ions yields the isotopes 247Es (einsteinium) or 260Db (dubnium), respectively. Furthermore, the element berkelium (243Bk isotope) had been first intentionally produced and identified by bombarding 241Am with alpha particles, in 1949, by the same Berkeley group, using the same 60 inch cyclotron. Similarly, nobelium was produced at the Joint Institute for Nuclear Research, Dubna, Russia, in 1965 in several reactions, one of which included irradiation of 243Am with 15N ions. Besides, one of the synthesis reactions for lawrencium, discovered by scientists at Berkeley and Dubna, included bombardment of 243Am with 18O.
Americium - 241
has been used as a portable source of both gamma rays and alpha
particles for a number of medical and industrial uses. The 60-keV gamma
ray emissions from 241Am in such sources can be used for indirect analysis of materials in radiography and X-ray fluorescence spectroscopy, as well as for quality control in fixed nuclear density gauges and nuclear densometers. For example, the element has been employed to gauge glass thickness to help create flat glass. Americium - 241
is also suitable for calibration of gamma ray spectrometers in the
low energy range, since its spectrum consists of nearly a single peak
and negligible Compton continuum (at least three orders of magnitude
lower intensity). Americium - 241
gamma rays were also used to provide passive diagnosis of thyroid
function. This medical application is however obsolete.
As a highly radioactive element, americium and its compounds must be handled only in an appropriate laboratory under special arrangements. Although most americium isotopes predominantly emit alpha particles which can be blocked by thin layers of common materials, many of the daughter products emit gamma rays and neutrons which have a long penetration depth.
If consumed, americium is excreted within a few days and only 0.05% is absorbed in the blood. From there, roughly 45% of it goes to the liver and 45% to the bones, and the remaining 10% is excreted. The uptake to the liver depends on the individual and increases with age. In the bones, americium is first deposited over cortical and trabecular surfaces and slowly redistributes over the bone with time. The biological half-life of 241Am is 50 years in the bones and 20 years in the liver, whereas in the gonads (testicles and ovaries) it remains permanently; in all these organs, americium promotes formation of cancer cells as a result of its radioactivity.
Americium often enters landfills from discarded smoke detectors.
The rules associated with the disposal of smoke detectors are relaxed
in most jurisdictions. In the U.S., the "Radioactive Boy Scout" David Hahn was
able to concentrate americium from smoke detectors after managing to
buy a hundred of them at remainder prices and also stealing a few. There have been cases of humans being contaminated with americium, the worst case being that of Harold McCluskey,
who at the age of 64 was exposed to 500 times the occupational standard
for americium - 241 as a result of an explosion in his lab. McCluskey
died at the age of 75, not as a result of exposure, but of a heart disease which he had before the accident.

Glenn Theodore Seaborg (Swedish: Glenn Teodor Sjöberg; April 19, 1912 – February 25, 1999) was an American scientist who won the 1951 Nobel Prize in Chemistry for "discoveries in the chemistry of the transuranium elements", contributed to the discovery and isolation of ten elements, and developed the actinide concept, which led to the current arrangement of the actinide series in the periodic table of the elements. He spent most of his career as an educator and research scientist at the University of California, Berkeley, where he became the second Chancellor in its history and served as a University Professor. Seaborg advised ten presidents from Harry S. Truman to Bill Clinton on nuclear policy and was the chairman of the United States Atomic Energy Commission from 1961 to 1971 where he pushed for commercial nuclear energy and peaceful applications of nuclear science. Throughout his career, Seaborg worked for arms control. He was signatory to the Franck Report and contributed to the achievement of the Limited Test Ban Treaty, the Nuclear Non - Proliferation Treaty, and the Comprehensive Test Ban Treaty. Seaborg was a well known advocate of science education and federal funding for pure research. He was a key contributor to the report "A Nation at Risk" as a member of President Reagan's National Commission on Excellence in Education and was the principal author of the Seaborg Report on academic science issued in the closing days of the Eisenhower administration.
Seaborg was the principal or co-discoverer of ten elements: plutonium, americium, curium, berkelium, californium, einsteinium, fermium, mendelevium, nobelium and element 106, which was named seaborgium in
his honor while he was still living. He also developed more than 100
atomic isotopes, and is credited with important contributions to the
chemistry of plutonium, originally as part of the Manhattan Project where he developed the extraction process used to isolate the plutonium fuel for the second atomic bomb.
Early in his career, Seaborg was a pioneer in nuclear medicine and
developed numerous isotopes of elements with important applications in
the diagnosis and treatment of diseases, most notably iodine - 131,
which
is used in the treatment of thyroid disease. In addition to his
theoretical work in the development of the actinide concept which placed
the actinide series beneath the lanthanide series on the periodic
table, Seaborg proposed the placement of super heavy elements in the transactinide and superactinide series. After sharing the 1951 Nobel Prize in Chemistry with Edwin McMillan, he received approximately 50 honorary doctorates and numerous other awards and honors. The list of things named after Seaborg ranges from his atomic element to an asteroid. Seaborg was a prolific author, penning more than 50 books and 500 journal articles, often in collaboration with others. He received so many awards and honors that he was once listed in the Guinness Book of World Records as the person with the longest entry in Who's Who in America.
Of Swedish, distant German and Belgian (Flemish and Walloon) ancestry, Seaborg was born in Ishpeming, Michigan, the son of Herman Theodore (Ted) and Selma Olivia Erickson Seaborg. He had one sister, Jeanette. When Glenn Seaborg was a boy, the family moved to the Seaborg Home in a subdivision called Home Gardens, that was later annexed to the City of South Gate, California, a suburb of Los Angeles.
He kept a daily journal from 1927 until he suffered a stroke in 1998. As a youth, Seaborg was both a devoted sports fan and an avid movie buff. His mother encouraged him to become a book keeper as she felt his literary interests were impractical. He did not take an interest in science until his junior year when he was inspired by Dwight Logan Reid, a chemistry and physics teacher at David Starr Jordan High School in Watts.
He graduated from Jordan in 1929 at the top of his class and received a bachelor's degree in chemistry at the University of California, Los Angeles, in 1933. While at UCLA, he was invited by his German professor to meet Albert Einstein,
an experience that had a profound impact on Seaborg and served as a
model of graciousness for his encounters with aspiring students in later
years. Seaborg worked his way through school as a stevedore (longshoreman), fruit packer and laboratory assistant.
Seaborg took his Ph.D. in chemistry at the University of California, Berkeley, in 1937 with a doctoral thesis on the inelastic scattering of neutrons in which he coined the term "nuclear spallation". He was a member of the professional chemistry fraternity Alpha Chi Sigma. As a graduate student in the 1930s Seaborg performed wet chemistry research for his advisor Gilbert Newton Lewis and published three papers with him on the theory of acids and bases. Seaborg then studied thoroughly the text Applied Radiochemistry by Otto Hahn, of the Kaiser Wilhelm Institute for Chemistry in Berlin and it had a major impact on his developing interests as a research scientist. For several years, Seaborg conducted important research in artificial radioactivity using the Lawrence cyclotron at UC Berkeley. He was excited to learn from others that nuclear fission was possible — but also chagrined, as his own research might have led him to the same discovery.
Seaborg also became expert in dealing with noted Berkeley physicist Robert Oppenheimer.
Oppenheimer had a daunting reputation, and often answered a junior
man's question before it had even been stated. Often the question
answered was more profound than the one asked, but of little practical
help. Seaborg learned to state his questions to Oppenheimer quickly and
succinctly.
Seaborg remained at the University of California, Berkeley for post doctoral research. He followed Frederick Soddy's work investigating isotopes and contributed to the discovery of more than 100 isotopes of elements. Using one of Lawrence's advanced cyclotrons, John Livingood, Fred Fairbrother, and Seaborg created a new isotope of iron, iron - 59 (Fe-59) in 1937. Iron - 59 was useful in the studies of the hemoglobin in human blood. In 1938, Livingood and Seaborg collaborated (as they did for five years) to create an important isotope of iodine, iodine - 131 (I-131) which is still used to treat thyroid disease. (Many years later, it was credited with prolonging the life of Seaborg's mother.) As a result of these and other contributions, Seaborg is regarded as a pioneer in nuclear medicine and is one of its most prolific discoverers of isotopes.
In 1939 he became an instructor in chemistry at Berkeley, was promoted to assistant professor in 1941 and professor in 1945.
UC Berkeley physicist Edwin McMillan had led a team that discovered element 93, neptunium in 1940. In November 1940, McMillan was persuaded to leave Berkeley temporarily to assist with urgent research in radar technology. Since Seaborg and his colleagues had perfected McMillan's oxidation - reduction technique for isolating neptunium, he asked McMillan for permission to continue the research and search for element 94. McMillan agreed to the collaboration. Seaborg first reported alpha decay proportionate to only a fraction of the element 93 under observation. The first hypothesis for this alpha particle accumulation was contamination by uranium, which produces alpha decay particles; analysis of alpha decay particles ruled this out. Seaborg then postulated that a distinct alpha producing element was being formed from element 93. In February 1941, Seaborg and his collaborators produced plutonium - 239 through the bombardment of uranium. This experimental achievement changed the course of human history in ways more profound than they could have ever imagined: the production of plutonium - 239 was successful. In their experiments bombarding uranium with deuterons, they observed the creation of neptunium, element 93. But it then underwent beta decay, forming a new element, plutonium, with 94 protons. Plutonium is fairly stable, but undergoes alpha decay, which explained the presence of alpha particles coming from neptunium. Thus, on March 28, 1941, Dr. Seaborg, physicist Emilio Segrè and Berkeley chemist Joseph W. Kennedy were able to show that plutonium (then known only as element 94239) underwent fission with slow neutrons, an important distinction that was crucial to the decisions made in directing Manhattan Project research. Room 307 of Gilman Hall on the campus at the University of California, Berkeley, where Seaborg did his work, has since been declared a U.S. National Historic Landmark.
In
addition to plutonium, he is credited as a lead discoverer of
americium, curium, and berkelium, and as a co-discoverer of californium,
einsteinium, fermium, mendelevium, nobelium and seaborgium. He shared
the Nobel Prize in Chemistry in 1951 with Edwin McMillan for
"their discoveries in the chemistry of the first transuranium
elements." He obtained patents on americium and curium, which were
developed in 1944 in Chicago at the wartime metallurgical laboratory
during the Manhattan project. His research contributions to all of the
other elements were conducted at the University of California, Berkeley.
On April 19, 1942, Seaborg reached Chicago, and joined the chemistry group at the Metallurgical Laboratory of the Manhattan Project at the University of Chicago, where Enrico Fermi and his group would later convert U238 to plutonium in the world's first controlled nuclear chain reaction using a chain reacting pile. Seaborg's role was to figure out how to extract the tiny bit of plutonium from the mass of uranium. Plutonium - 239 was isolated in visible amounts using a transmutation reaction on August 20, 1942 and weighed on September 10, 1942 in Seaborg's Chicago laboratory. He was responsible for the multi - stage chemical process that separated, concentrated and isolated plutonium. This process was further developed at the Clinton Engineering Works in Oak Ridge, Tennessee, and then entered full scale production at the Hanford Engineer Works, in Richland, Washington.
Seaborg's theoretical development of the actinide concept resulted in a redrawing of the Periodic Table of the Elements into its current configuration with the actinide series appearing below the lanthanide series. Seaborg developed the chemical elements americium and curium while
in Chicago. He managed to secure patents for both elements. His patent
on curium never proved commercially viable because of the element's
short half-life. Americium is commonly used in household smoke
detectors, however, and thus provided a good source of royalty income to
Seaborg in later years. Prior to the test of the first nuclear weapon,
Seaborg joined with several other leading scientists in a written
statement known as the Franck Report (secret
at the time but since published) calling on President Truman to conduct
a public demonstration of the atomic bomb witnessed by the Japanese
rather than engaging in a surprise attack. Truman instead proceeded to
drop two bombs, credited by most observers at the time with ending the
war, a uranium bomb on Hiroshima and a plutonium bomb on Nagasaki.
After the conclusion of World War II and the Manhattan Project, Seaborg was eager to return to academic life and university research free from the restrictions of wartime secrecy. In 1946, he added to his responsibilities as a professor by heading the nuclear chemistry research at the Lawrence Radiation Laboratory operated by the University of California on behalf of the United States Atomic Energy Commission. Seaborg was named one of the "Ten Outstanding Young Men in America" by the U.S. Junior Chamber of Commerce in 1947 (along with Richard Nixon and others). Seaborg was elected to the National Academy of Sciences in 1948. From 1954 to 1961 he served as associate director of the radiation laboratory. He was appointed by President Truman to serve as a member of the General Advisory Committee of the Atomic Energy Commission, an assignment he retained until 1960.
Seaborg served as chancellor at University of California, Berkeley, from
1958 to 1961. His term as Chancellor came at a time of considerable
controversy during the time of the free speech movement. In October
1958, he announced that the University had relaxed its prior
prohibitions on political activity on a test basis. Seaborg served on the Faculty Athletic Committee for several years and is the co-author of a book concerning the Pacific Coast Conference scandal and the founding of the Pac-10 (formerly
Pac-8), in which he played a role. Seaborg served on the President's
Science Advisory Commission during the Eisenhower administration, which
produced the report "Scientific Progress, the Universities, and the
Federal Government," also known as the "Seaborg Report," in November
1960. The Seaborg Report is credited with influencing the federal policy
towards academic science for the next eight years. In 1959, he helped
found the Berkeley Space Sciences Laboratory with UC president Clark Kerr.
After appointment by President John F. Kennedy and confirmation by the United States Senate, Seaborg was chairman of the United States Atomic Energy Commission (AEC) from 1961 to 1971. His pending appointment by President Kennedy was nearly derailed in late 1960 when members of the Kennedy transition team learned that Seaborg had been listed in a U.S. News and World Report article as a member of "Nixon's Brain Trust." Seaborg said that as a lifetime Democrat he was baffled when the article appeared associating him with Vice President Nixon, whom he considered a casual acquaintance.
While chairman of the AEC, Seaborg participated on the negotiating team for the Limited Test Ban Treaty (LTBT). Seaborg considered his contributions to the achievement of the LTBT as his greatest accomplishment. Despite strict rules from the Soviets about photography at the signing ceremony, Seaborg sneaked a tiny camera past the Soviet guards to take a close-up photograph of Soviet Premier Nikita Khrushchev as he signed the treaty.
Seaborg
was ardent supporter of large scale massive nuclear plants for
electricity generation despite concerns by industry insiders that such
large plants were vulnerable in that their nuclear cores could not be
properly contained in the event of an accident or operating emergency.
Seaborg received a letter dated August 16, 1966 from industry engineers
expressing these concerns at the time of the licensing of New York's
Indian Point reactor. This letter advised Seaborg and other AEC senior
members of these containment concerns which would later become known as
the "China Syndrome" resulting from uncontained core meltdowns. Seaborg
directed this letter not be released to the public as he feared it would
be misunderstood and therefore damage the nuclear industry in the
public's view even though the law required such letters be released for
public disclosure. This disclosure first came to light for public view
in the BBC documentary series, "Pandora's Box, A Is For Atom" dealing
with the early history of commercial nuclear development.
Seaborg enjoyed a close relationship with President Lyndon Johnson and influenced the administration to pursue the Nuclear Non - Proliferation Treaty.
Seaborg was called to the White House in the first week of the Nixon Administration in January 1969 to advise President Richard Nixon on his first diplomatic crisis involving the Soviets and nuclear testing. Seaborg clashed with Nixon presidential adviser John Ehrlichman over the treatment of a Jewish scientist whom the Nixon administration suspected of leaking nuclear secrets to Israel.
Seaborg published several books and journal articles during his tenure at the Atomic Energy Commission. His predictions concerning development of stable super heavy elements are considered among his most important theoretical contributions. Seaborg theorized the transactinide series and the superactinide series of undiscovered synthetic elements. While most of these theoretical future elements have extremely short half-lives and thus no expected practical applications, Seaborg theorized an island of stability for isotopes of certain elements.
When
Seaborg resigned as chairman of the Atomic Energy Commission in 1971,
he had served longer than any other Kennedy appointee.
Following his service as Chairman of the Atomic Energy Commission, Seaborg returned to UC Berkeley where he was awarded the position of University Professor. At the time, there had been fewer University Professors at UC Berkeley than Nobel prize winners. He also served as Chairman of the Lawrence Hall of Science. Seaborg served as President of the American Association for the Advancement of Science in 1972 and as President of the American Chemical Society in 1976. In 1976, when the Swedish king visited the United States, Seaborg played a major role in welcoming the Swedish Royal Family.
In 1980, he transmuted several thousand atoms of bismuth into gold at the Lawrence Berkeley Laboratory. His experimental technique, using nuclear physics, was able to remove protons and neutrons from the bismuth atoms. Seaborg's technique would have been far too expensive to enable routine manufacturing of gold, but his work is the closest to the mythical Philosopher's Stone.
In 1983, President Ronald Reagan appointed Seaborg to serve on the National Commission on Excellence in Education. Upon seeing the final draft report, Seaborg is credited with making comments that it was far too weak and did not communicate the urgency of the current crisis. He compared the crisis in education to the arms race, and stated that we are "a nation at risk." These comments led to a new introduction to the report and gave the report the famous title which focused national attention on education as an issue germane to the federal government.
Seaborg lived most of his later life in Lafayette, California, where he devoted himself to editing and publishing the journals that documented both his early life and later career. He rallied a group of scientists who criticized the science curriculum in the State of California which he viewed as far too socially oriented and not nearly focused enough on hard science. California Governor Pete Wilson appointed Seaborg to head a committee that proposed sweeping changes to California's science curriculum despite outcries from labor organizations and others.
On August 24, 1998, while in Boston to attend a meeting by the American Chemical Society, Seaborg suffered a stroke, which led to his death six months later on February 25, 1999 at his home in Lafayette.
During
his lifetime, Seaborg is said to have been the author or co-author of
more than 50 books and 500 scientific journal articles, many of them
brief reports on fast breaking discoveries in nuclear science while
other subjects, most notably the actinide concept, represented major
theoretical contributions in the history of science. He held more than 40 patents — among them the only patents ever issued for chemical elements, americium and curium.
He is also said to have received more than 50 degrees and honorary
degrees in his lifetime. At one time, he was listed in the Guinness Book of World Records as having the longest entry in Marquis Who's Who in America. In February 2005, Seaborg was posthumously inducted into the National Inventors Hall of Fame.
In 1942, Seaborg married Helen Griggs, the secretary of Ernest Lawrence.
Under wartime pressure, Seaborg had moved to Chicago while engaged to Griggs. When Seaborg returned to accompany Griggs for the journey back to Chicago, friends expected them to marry in Chicago. But, eager to be married, Seaborg and Griggs impulsively got off the train in the town of Caliente, Nevada, for what they thought would be a quick wedding. When they asked for City Hall, they found Caliente had none — they would have to travel 25 miles north to Pioche, the county seat. With no car, this was no easy feat but, happily, one of Caliente's newest deputy sheriffs turned out to be a recent graduate of the Cal Berkeley chemistry department and was more than happy to do a favor for Seaborg. The deputy sheriff arranged for the wedding couple to ride up and back to Pioche in a mail truck. The witnesses at the Seaborg wedding were a clerk and a janitor.
Glenn Seaborg and Helen Griggs Seaborg had six children, of whom the first, Peter Glenn Seaborg, died in 1997. The others were Lynne Seaborg Cobb, David Seaborg, Steve Seaborg, Eric Seaborg, and Dianne Seaborg.
Seaborg was an avid hiker. Upon becoming Chairman of the Atomic Energy Commission in 1961, he commenced taking daily hikes through a trail which he blazed at the headquarters site in Germantown, Maryland. He frequently invited colleagues and visitors to accompany him and the trail became known as the "Glenn Seaborg Trail."
He and his wife Helen are credited with blazing a 12 mile trail in the East Bay area near their Lafayette, California, home. This trail has since become a part of the American Hiking Association's cross country network of trails. Seaborg and his wife walked the trail network from Contra Costa County all the way to the California - Nevada border.
Seaborg was honored as Swedish - American of the Year in 1962 by the Vasa Order of America. In 1991, the organization named "Local Lodge Glenn T. Seaborg No. 719" in his honor during the Seaborg Honors ceremony at which he appeared. This lodge maintains a scholarship fund in his name, as does the unrelated Swedish - American Club of Los Angeles.
Seaborg kept a close bond to his Swedish origin. He visited Sweden every so often and his family were members of the Swedish Pemer Genealogical Society, a family association open for every descendant of the Pemer family, a Swedish family with German origin, from which Seaborg was descended on his mother's side.
He was elected a foreign member of the Royal Swedish Academy of Sciences in 1972 and the Royal Society of London.
Seaborg was an enthusiastic supporter of Cal's sports teams. San Francisco columnist Herb Caen was fond of pointing out that Seaborg's surname is an anagram of "Go Bears", a popular cheer at UC Berkeley.
The element seaborgium was named after Seaborg by Albert Ghiorso, E. Kenneth Hulet, and others, who also credited Seaborg as a co-discoverer. It was so named while Seaborg was still alive, which proved controversial. He influenced the naming of so many elements that with the announcement of seaborgium, it was noted in Discover magazine's review of the year in science that he could receive a letter addressed in chemical elements: seaborgium, lawrencium (for the Lawrence Berkeley Laboratory where he worked), berkelium, californium, americium.
While
it has been commonly stated that seaborgium is the only element to have
been named after a living person, this is not entirely accurate; both einsteinium and fermium were proposed as names of new elements discovered by Albert Ghiorso while Enrico Fermi and Albert Einstein were still living. The discovery of these elements and their names were kept secret under Cold War era
nuclear secrecy rules, however, and thus the names were not known by
the public or the broader scientific community until after the deaths of
Fermi and Einstein. Thus seaborgium is the only element to have been
publicly named after a living person.

Albert Ghiorso (July 15, 1915 – December 26, 2010) was an American nuclear scientist and co-discoverer of a record 12 chemical elements on the periodic table. His research career spanned five decades, from the early 1940s to the late 1990s.
Ghiorso was born in California July 15, 1915. He grew up in Alameda, California. As a teenager, he built radio circuitry and earned a reputation for establishing radio contacts at distances that outdid the military.
He received his BS in electrical engineering from the University of California, Berkeley, in 1937. After graduation, he worked for Reginald Tibbets, a prominent amateur radio operator who operated a business supplying radiation detectors to the government. Ghiorso's ability to develop and produce these instruments, as well as a variety of electronic tasks, brought him into contact with the nuclear scientists at the University of California Radiation Laboratory at Berkeley, in particular Glenn Seaborg. During a job in which he was to install an intercom at the lab, he met two secretaries, one of whom married Seaborg and the other, Wilma Belt, who became Albert's wife of 60+ years.
In
the early 1940s, Seaborg moved to Chicago to work on the Manhattan
Project. He invited Ghiorso to join him, and for the next four years
Ghiorso developed sensitive instruments for detecting the radiation
associated with nuclear decay, including spontaneous fission. One of
Ghiorso's breakthrough instruments was a 48 channel pulse height
analyzer, which enabled him to identify the energy, and therefore the
source, of the radiation. During this time they discovered two new
elements (95, americium and 96, curium), although publication was withheld until after the war.
After the war, Seaborg and Ghiorso returned to Berkeley, where they and colleagues used the 60" Crocker cyclotron to produce elements of increasing atomic number by bombarding exotic targets with helium ions. In experiments during 1949 - 1950, they produced and identified elements 97 (berkelium) and 98 (californium). In 1953, in a collaboration with Argonne Lab, Ghiorso and collaborators sought and found elements 99 (einsteinium and 100 (fermium), identified by their characteristic radiation in dust collected by airplanes from the first thermonuclear explosion (the Mike test). In 1955, the group used the cyclotron to produce 17 atoms of element 101 (mendelevium), the first new element to be discovered atom - by - atom. The recoil technique invented by Ghiorso was crucial to obtaining an identifiable signal from individual atoms of the new element.
In the mid 1950s it became clear that to extend the periodic chart any further, a new accelerator would be needed, and the Berkeley Heavy Ion Linear Accelerator (HILAC) was built, with Ghiorso in charge. That machine was used in the discovery of elements 102 - 106 (102, nobelium; 103, lawrencium; 104, rutherfordium; 105, dubnium and 106, seaborgium), each produced and identified on the basis of only a few atoms. The discovery of each successive element was made possible by the development of innovative techniques in robotic target handling, fast chemistry, efficient radiation detectors, and computer data processing. The 1972 upgrade of the HILAC to the superHILAC provided higher intensity ion beams, which was crucial to producing enough new atoms to enable detection of element 106.
With increasing atomic number, the experimental difficulties of producing and identifying a new element increase significantly. In the 1970s and 1980s, resources for new element research at Berkeley were diminishing, but the GSI laboratory at Darmstadt, Germany, under the leadership of Peter Armbruster and with considerable resources, was able to produce and identify elements 107 - 109 (107, bohrium; 108, hassium and 109, meitnerium). In the early 1990s, the Berkeley and Darmstadt groups made a collaborative attempt to create element 110. Experiments at Berkeley were unsuccessful, but eventually elements 110 - 112 (110, darmstadtium; 111, roentgenium and 112, copernicium) were identified at the Darmstadt laboratory. Subsequent work at the JINR laboratory at Dubna, led by Yuri Oganessian, was successful in identifying elements 113 - 118 (113, ununtrium; 114, ununquadium; 115, ununpentium; 116,ununhexium; 117, ununseptium and 118, ununoctium), thereby completing the seventh row of the periodic table of the elements.
Ghiorso invented numerous techniques and machines for isolating and identifying heavy elements atom - by - atom. He is generally credited with implementing the multichannel analyzer and the technique of recoil to isolate reaction products, although both of these were significant extensions of previously understood concepts. His concept for a new type of accelerator, the Omnitron, is acknowledged to have been a brilliant advance that probably would have enabled the Berklely lab to discover numerous additional new elements, but the machine was never built, a victim of the evolving political landscape of the 1970s in the U.S. that de-emphasized basic nuclear research and greatly expanded research on environmental, health, and safety issues. Partially as a result of the failure to build the Omnitron, Ghiorso (together with colleagues Bob Main and others) conceived the joining of the HILAC and the Bevatron, which he called the Bevalac. This combination machine, an ungainly articulation across the steep slope at the Rad Lab, provided heavy ions at GeV energies, thereby enabling development of two new fields of research: "high energy nuclear physics," meaning that the compound nucleus is sufficiently hot to exhibit collective dynamical effects, and heavy ion therapy, in which high energy ions are used to irradiate tumors in cancer patients. Both of these fields have expanded into activities in many laboratories and clinics world wide.
In
his later years, Ghiorso continued research toward finding superheavy
elements, fusion energy, and innovative electron beam sources. He was a
non - participating co-author of the experiments in 1999 that gave
evidence of elements 116 and 118. He also had brief research interests
in the free quark experiment of William Fairbank of Stanford, in the
discovery of element 43, the electron disk accelerator, among others.
Albert Ghiorso is credited with having co-discovered the following elements
- Americium ca. 1945 (element 95)
- Curium in 1944 (element 96)
- Berkelium in 1949 (element 97)
- Californium in 1950 (element 98)
- Einsteinium in 1952 (element 99)
- Fermium in 1953 (element 100)
- Mendelevium in 1955 (element 101)
- Nobelium in 1958–59 (element 102)
- Lawrencium in 1961 (element 103)
- Rutherfordium in 1969 (element 104)
- Dubnium in 1970 (element 105)
- Seaborgium in 1974 (element 106)
Ghiorso personally selected some of the names recommended by his group for the new elements. His original name for element 105 (hahnium) was changed by the International Union of Pure and Applied Chemistry (IUPAC) to dubnium, to recognize the contributions of the laboratory at Dubna, Russia, in the search for trans - fermium elements. His recommendation for element 106, seaborgium, was accepted only after extensive debate about naming an element after a living person. In 1999, evidence for two superheavy elements (element 116 and element 118) was published by a group in Berkeley. The discovery group intended to propose the name ghiorsium for element 118, but eventually the data were found to have been tampered and in 2002 the claims were withdrawn. Ghiorso's lifetime output comprised about 170 technical papers, most published in The Physical Review.
Ghiorso is famous among his colleagues for his endless stream of creative "doodles," which define an art form suggestive of fractals. He also developed a state - of - the - art camera for birdwatching, and was a constant supporter of environmental causes and organizations.