<Back to Index>
- Element 97 Berkelium Bk, 1949
- Chemist Glenn Theodore Seaborg, 1912
- Chemist Albert Ghiorso, 1915
- Chemist Stanley Gerald Thompson, 1912
PAGE SPONSOR
Berkelium is a transuranic radioactive chemical element with the symbol Bk and atomic number 97, a member of the actinide and transuranium element series. It is named after the city of Berkeley, California, the location of the University of California Radiation Laboratory where it was discovered in December 1949. This was the fifth transuranium element discovered after neptunium, plutonium, curium and americium.
The major isotope of berkelium, berkelium - 249, is synthesized in minute quantities in dedicated high flux nuclear reactors, mainly at the Oak Ridge National Laboratory in Tennessee, USA, and at the Research Institute of Atomic Reactors in Dimitrovgrad, Russia. The production of the second important isotope berkelium - 247 involves the irradiation of the rare synthetic isotope curium - 244 with high energy alpha particles.
Just over one gram of berkelium has been produced in the United States since 1967. There is no practical application of berkelium outside of scientific research which is mostly directed at the synthesis of heavier transuranic elements and transactinides. A 22 milligram batch of berkelium - 249 was prepared during a 250 day irradiation period and then purified for a further 90 days at Oak Ridge in 2009. This sample was used to synthesize the element ununseptium for the first time in 2009 at the Joint Institute for Nuclear Research, Russia, after it was bombarded with calcium - 48 ions for 150 days. This was a culmination of the Russia — US collaboration on the synthesis of elements 113 to 118.
Berkelium is a soft, silvery white, radioactive metal. The berkelium - 249 isotope emits low energy electrons and thus is relatively safe to handle. However, it decays with a half-life of 330 days to californium - 249,
which is a strong and hazardous emitter of alpha particles. This
gradual transformation is an important consideration when studying the
properties of elemental berkelium and its chemical compounds, since the
formation of californium brings not only chemical contamination, but
also self radiation damage, and self heating from the emitted alpha
particles.
Although very small amounts of berkelium were possibly produced in previous nuclear experiments, it was first intentionally synthesized, isolated and identified in December 1949 by Glenn T. Seaborg, Albert Ghiorso and Stanley G. Thompson. They used the 60 inch cyclotron at the University of California, Berkeley. Similar to the nearly simultaneous discovery of americium (element 95) and curium (element 96) in 1944, the new elements berkelium and californium (element 98) were both produced in 1949 – 1950.
The name choice for element 97 followed the previous tradition of the Californian group to draw an analogy between the newly discovered actinide and the lanthanide element positioned above it in the periodic table. Previously, americium was named after a continent as its analogue europium, and curium honored scientists Marie and Pierre Curie as the lanthanide above it, gadolinium, was named after the explorer of the rare earth elements Johan Gadolin. Thus the discovery report by the Berkeley group reads: "It is suggested that element 97 be given the name berkelium (symbol Bk) after the city of Berkeley in a manner similar to that used in naming its chemical homologue terbium (atomic number 65) whose name was derived from the town of Ytterby, Sweden, where the rare earth minerals were first found." This tradition ended on berkelium though as the naming of the next discovered actinide, californium, was not related to its lanthanide analogue dysprosium, but after the discovery place.
The most difficult steps in the synthesis of berkelium were its separation from the final products and the production of sufficient quantities of americium for the target material. First, americium (241Am) nitrate solution was coated on a platinum foil, the solution was evaporated and the residue converted by annealing to americium dioxide (AmO2). This target was irradiated with 35 MeV alpha particles for 6 hours in the 60 inch cyclotron at the Lawrence Radiation Laboratory, University of California, Berkeley. The (α,2n) reaction induced by the irradiation yielded the 243Bk isotope and two free neutrons:
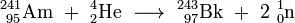
After the irradiation, the coating was dissolved with nitric acid and then precipitated as the hydroxide using concentrated aqueous ammonia solution. The product was centrifugated and re-dissolved in nitric acid. To separate berkelium from the unreacted americium, this solution was added to a mixture of ammonium and ammonium sulfate and heated to convert all the dissolved americium into the oxidation state +6. Unoxidized residual americium was precipitated by the addition of hydrofluoric acid as americium (III) fluoride (AmF3). This step yielded a mixture of the accompanying product curium and the expected element 97 in form of trifluorides. The mixture was converted to the corresponding hydroxides by treating it with potassium hydroxide, and after centrifugation, was dissolved in perchloric acid.
Further separation was carried out in the presence of a citric acid / ammonium buffer solution in a weakly acidic medium (pH ≈ 3.5), using ion exchange at elevated temperature. The chromatographic separation
behavior was then unknown for the element 97, but was anticipated by
analogy with terbium (elution curves). First results were
disappointing as no alpha particle emission signature could be detected
from the elution product. Only the further search for characteristic X-rays and conversion electron signals resulted in the identification of a berkelium isotope. Its mass number was uncertain between 243 and 244 in the initial report, but was later established as 243.
Berkelium is a synthetic, silvery white, radioactive actinide metal. In the periodic table, it is located right to the actinide curium, left to the actinide californium and below the lanthanide terbium with which it shares many similarities in physical and chemical properties. Its density of 14.78 g/cm3 lies between those of curium (13.52 g/cm3) and californium (15.1 g/cm3), as does its melting point of 986 °C, below that of curium (1340 °C) but higher than that of californium (900 °C). Berkelium is relatively soft and has one of the lowest bulk moduli among the actinides, at about 20 GPa (2×1010Pa).
Berkelium (III) ions show two sharp fluorescence peaks at 652 nanometers (red light) and 742 nanometers (deep red – near infrared) due to internal transitions at the f-electron shell. The relative intensity of these peaks depends on the excitation power and temperature of the sample. This emission can be observed, for example, after dispersing berkelium ions in a silicate glass, by melting the glass in presence of berkelium oxide or halide.
Between 70 K and room temperature, berkelium behaves as a Curie – Weiss paramagnetic material with an effective magnetic moment of 9.69 Bohr magnetons (µB) and a Curie temperature of 101 K. This magnetic moment is almost equal to the theoretical value of 9.72 µB calculated within the simple atomic L-S coupling model. Upon cooling to about 34 K, berkelium undergoes a transition to an antiferromagnetic state. Enthalpy of dissolution in hydrochloric acid at standard conditions is −600 kJ/mol−1, from which the standard enthalpy change of formation (ΔfH°) of aqueous Bk3+ ions is obtained as −601 kJ/mol−1. The standard potential Bk3+/Bk0 is −2.01 V. The ionization potential of a neutral berkelium atom is 6.23 eV.
At ambient conditions, berkelium assumes its most stable α form which has a hexagonal symmetry, space group P63/mmc, lattice parameters of 341 pm and 1107 pm. The crystal has a double hexagonal close packing structure with the layer sequence ABAC and so is isotypic with α-lanthanum and α-forms of actinides beyond curium. This crystal structure changes with pressure and temperature. When compressed at room temperature to 7 GPa, α-berkelium transforms to the beta modification, which has a face centered cubic (fcc) symmetry and space group Fm3m. This transition occurs without change in volume, but the enthalpy increases by 3.66 kJ/mol. Upon further compression to 25 GPa, berkelium transforms to an orthorhombic γ-berkelium structure similar to that of α-uranium. This transition is accompanied by a 12% volume decrease and delocalization of the electrons at the 5f electron shell. No further phase transitions are observed up to 57 GPa.
Upon heating, α-berkelium transforms into another "metastable" phase (that is at ambient temperature it gradually converts back to α-berkelium). This phase has an fcc lattice (but slightly different from β-berkelium), space group Fm3m and the lattice constant of 500 pm; this fcc structure is equivalent to the closest packing with the sequence ABC. The temperature of the phase transition is believed to be quite close to the melting point.
Like all actinides, berkelium dissolves in various aqueous inorganic acids, liberating gaseous hydrogen and converting into the berkelium (III) state. This trivalent oxidation state is the most stable, especially in aqueous solutions, but tetravalent and possibly divalent berkelium compounds are also known. The existence of divalent berkelium salts is uncertain and has only been reported in mixed lanthanum chloride - strontium chloride melts. A similar behavior is observed for the lanthanide analogue of berkelium, terbium. Aqueous solutions of Bk3+ ions are green in most acids. The color of the Bk4+ ions is yellow in hydrochloric acid and orange yellow in sulfuric acid. Berkelium does not react rapidly with oxygen at room temperature, possibly due to the formation of a protective oxide layer surface. However, it reacts with molten metals, hydrogen, halogens, chalcogens and pnictogens to form various binary compounds.
About twenty isotopes and six nuclear isomers (excited
states of an isotope) of berkelium have been characterized with the
atomic numbers ranging from 235 to 254. All of them are radioactive. The
longest half-lives are observed for 247Bk (1,380 years), 248Bk (9 years) and 249Bk
(330 days); the half-lives of the other isotopes range from
microseconds to several days. The isotope which is the easiest to
synthesize is berkelium - 249. This emits mostly soft β-particles which are inconvenient for detection. Its alpha radiation is rather weak – 1.45×10−3%
with respect to the β-radiation – but is sometimes used to detect this
isotope. The second important berkelium isotope, berkelium - 247, is an
alpha emitter as most actinide isotopes.
The longest lived isotope of berkelium (247Bk) has a half-life of only 1,380 years. Therefore, all primordial berkelium, that is berkelium present on the Earth during its formation, should have decayed by now. On Earth, berkelium is mostly concentrated in certain areas, which were used for the atmospheric nuclear weapons tests between 1945 and 1980, as well as at the sites of nuclear incidents, such as Chernobyl disaster, Three Mile Island accident and 1968 Thule Air Base B-52 crash. Analysis of the debris at the testing site of the first U.S. hydrogen bomb, Ivy Mike, (1 November 1952, Enewetak Atoll), revealed high concentrations of various actinides, including berkelium. For reasons of military secrecy, this result was published only in 1956.
Nuclear reactors produce mostly, among the berkelium isotopes, berkelium - 249. During the storage and before the fuel disposal, most of it breaks down to californium - 249. The latter has a long half-life, of 351 years, and is therefore undesirable in the disposal products.
A few atoms of berkelium can be produced by neutron capture reactions and beta decay in very highly concentrated uranium bearing deposits, thus making it the rarest naturally occurring element.
Berkelium is produced by bombarding lighter actinides uranium (238U) or plutonium (239Pu) with neutrons in a nuclear reactor. In a more common case of uranium fuel, plutonium is produced first by neutron capture (the so-called (n,γ) reaction or neutron fusion) followed by beta decay:
![\mathrm{^{238}_{\ 92}U\ \xrightarrow {(n,\gamma)} \ ^{239}_{\ 92}U\ \xrightarrow [23.5 \ min]{\beta^-} \ ^{239}_{\ 93}Np\ \xrightarrow [2.3565 \ d]{\beta^-} \ ^{239}_{\ 94}Pu}](aux2.png) (the times are half-lives)
(the times are half-lives)
Plutonium - 239 is further irradiated by a source that has a high neutron flux, several times higher than a conventional nuclear reactor, such as the 85 megawatt High Flux Isotope Reactor (HFIR) at the Oak Ridge National Laboratory in Tennessee, USA. The higher flux promotes fusion reactions involving not one but several neutrons, converting 239Pu to 244Cm and then to 249Cm:
![\mathrm{^{239}_{\ 94}Pu\ \xrightarrow {4(n,\gamma)} \ ^{243}_{\ 94}Pu\ \xrightarrow [4.956 \ h]{\beta^-} \ ^{243}_{\ 95}Am\ \xrightarrow {(n,\gamma)} \ ^{244}_{\ 95}Am\ \xrightarrow [10.1 \ h]{\beta^-} \ ^{244}_{\ 96}Cm} \quad; \quad \mathrm{^{244}_{\ 96}Cm\ \xrightarrow {5(n,\gamma)} \ ^{249}_{\ 96}Cm}](aux3.png)
Curium - 249 has a short half-life of 64 minutes, and thus its further conversion to 250Cm has a low probability. Instead, it transforms by beta decay into 249Bk:
![\mathrm{^{249}_{\ 96}Cm\ \xrightarrow [64.15 \ min]{\beta^-} \ ^{249}_{\ 97}Bk\ \xrightarrow [330 \ d]{\beta^-} \ ^{249}_{\ 98}Cf}](aux4.png)
The thus produced 249Bk has a long half-life of 330 days and thus can capture another neutron. However, the product, 250Bk, again has a relatively short half-life of 3.212 hours and thus, does not yield higher actinides. Instead decays to the californium isotope 250Cf:
![\mathrm{^{249}_{\ 97}Bk\ \xrightarrow {(n,\gamma)} \ ^{250}_{\ 97}Bk\ \xrightarrow [3.212 \ h]{\beta^-} \ ^{250}_{\ 98}Cf}](aux5.png)
The above reactions illustrate that although 247Bk is the most stable isotope of berkelium, its production in nuclear reactors is very inefficient. This makes 249Bk the most accessible isotope of berkelium, which still, is available only in small quantities (only 0.66 grams have been produced in the US over the period 1967 – 1983) at a high price of the order 185 USD per microgram.
The isotope 248Bk was first obtained in 1956 by bombarding a mixture of curium isotopes with 25 MeV α-particles. Although its direct detection was hindered by strong signal interference with 245Bk, the existence of a new isotope was proven by the growth of the decay product 248Cf which had been previously characterized. The half-life of 248Cf was estimated as 23 ± 5 hours and a more reliable value still is not known. Berkelium - 247 was produced during the same year by irradiating 244Cm with alpha particles:
![\mathrm{^{244}_{\ 96}Cm\ \xrightarrow[]{(\alpha,n)} \ ^{247}_{\ 98}Cf\ \xrightarrow[3,11 \ h]{\epsilon} \ ^{247}_{\ 97}Bk}](aux6.png)
![\mathrm{^{244}_{\ 96}Cm\ \xrightarrow[]{(\alpha,p)} \ ^{247}_{\ 97}Bk}](aux7.png)
Berkelium - 242 was synthesized in 1979 by bombarding 235U with 11B, 238U with 10B, 232Th with 14N or 232Th with 15N. It converts by electron capture to 242Cm with a half-life of 7.0 ± 1.3 minutes. A search for an initially suspected isotope 241Bk was then unsuccessful.
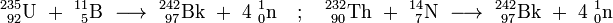
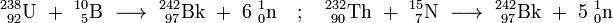
The fact that berkelium readily assumes oxidation state +4 in solids, and is relatively stable in this state in liquids greatly assists separation of berkelium away from many other actinides. These are inevitably produced in relatively large amounts during the nuclear synthesis and often favor the +3 state. This fact was not yet known in the initial experiments, which used a more complex separation procedure. Various oxidation agents can be applied to the berkelium (III) solutions to convert it to the +4 state, such as bromates (BrO3–), bismuthates (BiO3–), chromates (CrO42– and Cr2O72–), silver (I) thiolate (Ag2S2O8), lead (IV) oxide (PbO2), ozone (O3), or photochemical oxidation procedures. Berkelium (IV) is then extracted with ion exchange, extraction chromatography or liquid - liquid extraction using HDEHP (bis - (2 - ethylhexyl) phosphoric scid), amines, tributyl phosphate or various other reagents. These procedures separate berkelium from most trivalent actinides and lanthanides, except for the lanthanide cerium (lanthanides are absent in the irradiation target but are created in various nuclear fission decay chains).
A more detailed procedure adopted at the Oak Ridge National Laboratory was as follows: the initial mixture of actinides is processed with ion exchange using lithium chloride reagent, then precipitated as hydroxides, filtered and dissolved in nitric acid. It is then treated with high pressure elution from cation exchange resins, and the berkelium phase is oxidized and extracted using one of the procedures described above. Reduction
of the thus obtained berkelium (IV) to the +3 oxidation state yields a
solution, which is nearly free from other actinides (but contains
cerium). Berkelium and cerium are then separated with another round of
ion exchange treatment.
In order to characterize chemical and physical properties of solid berkelium and its compounds, a program was initiated in 1952 at the Material Testing Reactor, Arco, Idaho, US. It resulted in preparation of an eight gram plutonium - 239 target and in the first production of macroscopic quantities (0.6 micrograms) of berkelium by Burris B. Cunningham and Stanley G. Thompson in 1958, after a continuous reactor irradiation of this target for six years. This irradiation method was, and still is the only way of producing weighable amounts of the element, and most solid state studies of berkelium have been conducted on microgram or submicrogram sized samples.
The world's major irradiation sources are the 85 megawatt High Flux Isotope Reactor at the Oak Ridge National Laboratory in Tennessee, USA, and the SM-2 loop reactor at the Research Institute of Atomic Reactors (NIIAR) in Dimitrovgrad, Russia, which are both dedicated to the production of transcurium elements (atomic number greater than 96). These facilities have similar power and flux levels, and are expected to have comparable production capacities for transcurium elements, although the quantities produced at NIIAR are not widely reported. In a "typical processing campaign" at Oak Ridge, tens of grams of curium are irradiated to produce decigram quantities of californium, milligram quantities of berkelium - 249 and einsteinium, and picogram quantities of fermium. In total, just over one gram of berkelium - 249 has been produced at Oak Ridge since 1967.
The first berkelium metal sample weighing 1.7 micrograms was prepared in 1971 by the reduction of berkelium (III) fluoride with lithium vapor at 1000 °C; the fluoride was suspended on a tungsten wire above a tantalum crucible containing molten lithium. Later, metal samples weighting up to 0.5 milligrams were obtained with this method.
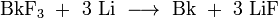
Similar results are obtained with berkelium (IV) fluoride. Berkelium metal can also be produced by the reduction of berkelium (IV) oxide with thorium or lanthanum.
Two oxides of berkelium are known, with the berkelium oxidation state of +3 (Bk2O3) and +4 (BkO2). Berkelium (IV) oxide is a brown solid that crystallizes in a cubic (fluorite) crystal structure. Berkelium (III) oxide is formed from BkO2 by reduction with molecular hydrogen:
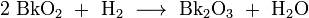
It is a yellow-green solid with a melting point of 1920 °C and a body centered cubic (bcc) crystal lattice. Upon heating to 1200 °C, the cubic Bk2O3 transforms to a monoclinic structure, which further converts to a hexagonal phase at 1750 °C, and the latter transition is reversible. Such three phase behavior is typical for the actinide sesquioxides. Berkelium (II) oxide, BkO, has been reported as a brittle gray solid with a face centered cubic (fcc) structure, but its exact chemical composition remains uncertain.
In halides, berkelium assumes the oxidation states +3 and +4. The +3 state is the most stable, especially in solutions, while the tetravalent halides BkF4 and Cs2BkCl6 are only known in the solid phase. The coordination of berkelium atom in its trivalent fluoride and chloride is tricapped trigonal prismatic, with the coordination number of 9. In trivalent bromide, it is bicapped trigonal prismatic (coordination 8) or octahedral (coordination 6), and in the iodide it is octahedral.
Berkelium (IV) fluoride (BkF4) is a yellow green ionic solid which crystallizes in the monoclinic crystal system and is isotypic with uranium tetrafluoride or zirconium (IV) fluoride.
Berkelium (III) fluoride (BkF3) is also a yellow green solid, but it has two crystalline structures. The most stable phase at low temperatures has an orthorhombic symmetry, isotypic with yttrium (III) fluoride. Upon heating to between 350 and 600 °C, it transforms to a trigonal structure found in lanthanum (III) fluoride.
Visible amounts of berkelium (III) chloride (BkCl3) were first isolated and characterized in 1962, and weighed only 3 billionths of a gram. It can be prepared by introducing hydrogen chloride vapors into an evacuated quartz tube containing berkelium oxide at a temperature about 500 °C. This green solid has a melting point of 600 °C, and crystallized in the hexagonal crystal system isotypic with uranium (III) chloride. Upon heating to nearly melting point, BkCl3 converts into an orthorhombic phase.
Two
forms of berkelium (III) bromide are known: a monoclinic one with
berkelium having coordination 6, and an orthorhombic one with
coordination 8. The latter is less stable and transforms to the former phase upon heating
to about 350 °C. An important phenomenon for radioactive solids
has been studied on these two crystal forms: the structure of fresh and
aged 249BkBr3 samples was probed by X-ray diffraction over
a period longer than 3 years, so that various fractions of
berkelium - 249 had converted to californium - 249. No change in structure
was observed upon 249BkBr3 — 249CfBr3 transformation, even though the orthorhombic bromide was previously unknown for
californium. However, other differences were noted for 249BkBr3 and 249CfBr3. For example, the latter could be reduced with hydrogen to 249CfBr2, but the former could be not – this result was reproduced on individual 249BkBr3 and 249CfBr3 samples, as well on the samples containing both bromides. The
intergrowth of californium in berkelium occurs at a rate of 0.22% per
day and is an intrinsic obstacle in studying berkelium properties.
Beside a chemical contamination, 249Cf,
as an alpha emitter brings undesirable self damage of the crystal
lattice and the resulting self heating. The chemical effect however can
be avoided by performing measurements as a function of time and
extrapolating the obtained results.
The pnictides of berkelium - 249 of the type BkX are known for the elements nitrogen, phosphorus, arsenic and antimony. They crystallize in the rock salt structure and are prepared by the reaction of either berkelium (III) hydride (BkH3) or metallic berkelium with these elements at elevated temperature (about 600 °C) under high vacuum.
Berkelium (III) sulfide, Bk2S3, was prepared by either treating berkelium oxide with a mixture of hydrogen sulfide and carbon disulfide vapors at 1130 °C, or by directly reacting metallic berkelium with elemental sulfur. These procedures yielded brownish black crystals with a cubic symmetry.
Berkelium (III) and berkelium (IV) hydroxides are both stable in 1 molar solutions of sodium hydroxide. Berkelium (III) phosphate (BkPO4) has been prepared as a solid, which shows strong fluorescence under excitation with a green light. Berkelium hydrides are produced by reacting metal with hydrogen gas at temperatures about 250 °C. They are non - stoichiometric with the nominal formula BkH2+x (0 < x < 1). Whereas the trihydride has a hexagonal symmetry, the dihydride crystallizes in an fcc structure. Several other salts of berkelium are known, including an oxysulfide (Bk2O2S), and hydrated nitrate (Bk(NO3)3·4H2O), chloride (BkCl3·6H2O), sulfate (Bk2(SO4)3·12H2O) and oxalate (Bk2(C2O4)3·4H2O). Thermal decomposition at about 600 °C in an argon atmosphere (to avoid oxidation to BkO2) of Bk2(SO4)3·12H2O yields the body centered orthorhombic crystals of berkelium (III) oxysulfate (Bk2O2SO4). This compound is thermally stable to at least 1000 °C in inert atmosphere.
Berkelium forms a trigonal (η5–C5H5)3Bk metallocene complex with three cyclopentadienyl rings, which can be synthesized by reacting berkelium (III) chloride with the molten beryllocene (Be(C5H5)2) at about 70 °C. It has an amber color, an orthorhombic symmetry and a density of 2.47 g/cm3.
The complex is stable to heating to at least 250 °C, and
sublimates without melting at about 350 °C. The high
radioactivity of berkelium gradually destroys the compound (within a
period of weeks). One cyclopentadienyl ring in (η5–C5H5)3Bk can be substituted by chlorine to yield [Be(C5H5)2Cl]2. The optical absorption spectra of this compound are very similar to those of (η5–C5H5)3Bk.
There is no use for any isotope of berkelium outside of basic scientific research. Berkelium - 249 is a common target nuclide to prepare still heavier transuranic elements and transactinides, such as lawrencium, rutherfordium and bohrium. It is also useful as a source of the isotope californium - 249, which is used for studies on the chemistry of californium in preference to the more radioactive californium - 252 that is produced in neutron bombardment facilities such as the HFIR.
A
22 milligram batch of berkelium - 249 was prepared in a 250 day
irradiation and then purified for 90 days at Oak Ridge in 2009. This
target yielded the first 6 atoms of element ununseptium at the Joint Institute for Nuclear Research (JINR), Dubna,
Russia, after bombarding it with calcium ions in the U400 cyclotron for
150 days. This synthesis was a culmination of the Russia — US
collaboration between JINR and Lawrence Livermore National Laboratory on the synthesis of elements 113 to 118 which was initiated in 1989.
The nuclear fission properties of berkelium are different from those of the neighboring actinides curium and californium, and they suggest berkelium to perform poorly as a fuel in a nuclear reactor. Specifically, berkelium - 249 has a moderately large neutron capture cross section of 710 barns for thermal neutrons, 1200 barns resonance integral, but very low fission cross section for thermal neutrons. In a thermal reactor, much of it will therefore be converted to berkelium - 250 which quickly decays to californium - 250. In principle, berkelium - 249 can sustain a nuclear chain reaction in a fast breeder reactor. Its critical mass is relatively high at 192 kg; it can be reduced with a water or steel reflector but would still exceed the world production of this isotope.
Berkelium - 247
can maintain chain reaction both in a thermal neutron and in a
fast neutron reactor, however, its production is rather complex and thus
the availability is much lower than its critical mass, which is about
75.7 kg for a bare sphere, 41.2 kg with a water reflector and
35.2 kg with a steel reflector (30 cm thickness).
Little is known about the effects of berkelium on human body, and analogies with other elements may not be drawn because of different radiation products (electrons for berkelium and alpha particles and/or neutrons for most other actinides). The low energy of electrons emitted from berkelium - 249 (less than 126 keV) hinders its detection, due to signal interference with other decay processes, but also makes this isotope relatively harmless to humans as compared to other actinides. However, berkelium - 249 transforms with a half-life of only 330 days to the strong alpha emitter californium - 249, which is rather dangerous and has to be handled in a glove box in a dedicated laboratory.
Most
available berkelium toxicity data originate from research on animals.
Upon ingestion by rats, only about 0.01% berkelium ends in the blood
stream. From there, about 65% goes to the bones, where it remains for
about 50 years, 25% to the lungs (biological half-life about
20 years), 0.035% to the testicles or 0.01% to the ovaries where
berkelium stays indefinitely. The balance of about 10% is excreted. In all these organs berkelium might promote cancer, and in the skeletal system its
radiation can damage red blood cells. The maximum permissible body
burden for the isotope berkelium - 249 in the human skeleton is
0.4 nanograms.

Glenn Theodore Seaborg (Swedish: Glenn Teodor Sjöberg; April 19, 1912 – February 25, 1999)

Albert Ghiorso (July 15, 1915 – December 26, 2010)

Stanley Gerald Thompson (1912 – 1976) was an American chemist. He discovered together with Glenn T. Seaborg several of the transuranium elements. He received a Guggenheim Fellowship (Natural Sciences - Chemistry) in 1954.